Si vous faites des simulations statistiques lourdes, vous devez ou devriez bénéficier de la parallélisation des ordinateurs multi-coeurs. Un quadri-coeur peut exécuter environ quatre fois plus vite (sauf saturation de la bande passante RAM) les simulations qu’un mono-coeur. Les outils de parallélisation tels que le package R « parallel » permettent de choisir le nombre de noeuds d’exécution. Quelle valeur choisir ?
La réponse est simple : choisissez autant de noeuds que votre processeur n’a de threads, ce qui est habituellement égal au double du nombre de coeurs. Vous pouvez éventuellement en mettre un ou deux de moins si vous voulez continuer à utiliser votre PC avec confort.
Exemple : processeur 4 coeurs et 8 threads.
Si vous utilisez quatre noeuds, vous gagnerez en performance d’un facteur quatre.
Si vous utilisez cinq noeuds, vous gagnerez en performance d’un facteur 4,2 environ par rapport à un seul noeud. Et vous gagnerez environ 0,2 unité de performance par noeud supplémentaire jusqu’à 8 noeuds. Avec 8 noeuds vous aurez un calcul 4+0.2×4=4.8 fois plus rapide qu’avec un seul noeud. Au delà, vous perdrez un peu en performance.
Limites de la règle n=threads
Parfois on peut gagner à mettre moins d’unité de calcul que ça, pour ne pas saturer la bande passante de la RAM lorsque les algorithmes travaillent sur de grandes quantité de données à la fois. Il est rare que vous ayez intérêt à en mettre moins que de coeurs.
Si on accède intensément à un disque dur, alors les accès alternés peuvent être très lent, à cause du temps d’accès long, et il peut être préférable de ne lancer qu’un seul noeud. Ce phénomène est moindre avec les Solid State Drives (SSD).
Pourquoi ?
Un coeur est un ensemble d’unités de calcul arithmétique et flottantes regroupés pour exécuter du code. Lorsqu’on utilise un coeur simplement, les unités de calcul ne sont pas saturées 100% du temps car certains calculs dépendent des précédents. Par exemple, si on calcule (5+3)×(2+8+4), on doit réaliser trois additions avant de réaliser la multiplication. Pendant que les unités de calcul d’addition sont utilisées, l’unité de multiplication est susceptible d’être au repos. Si on utilise deux threads sur le même coeur, un deuxième fil d’exécution est susceptible de saisir cette opportunité pour faire une multiplication dont elle a besoin à ce moment là. Néanmoins, avec deux threads sur un même coeur, ce dernier est presque en permanence à saturation et on ne gagne qu’environ 20% par rapport à une exécution séquentielle des simulations sur un seul thread.
Les détails sont plus complexes mais nécessitent de maîtriser l’architecture interne d’un PC. Une unité de calcul est plus ou moins spécialisée (p.e. Unité Arithmétique et Logique ou ALU en anglais).
Le tableau 1, dans les essais cliniques randomisés, décrit généralement les caractéristiques initiales des deux groupes randomisés. Âge, sexe, état général, comorbidités, sévérité de la maladie d’intérêt. Les grandes revues, telles que je New England Journal of Medicine présentent généralement les caractéristiques sous forme d’un tableau avec une colonne par groupe de randomisation. Il arrive, dans d’autres revues, que des comparaisons statistiques soient faites entre les deux groupes sous forme d’une dernière colonne de petits p. Quelle présentation est la plus pertinente ?
Résumé
Un petit p entre 0.01 et 0.05 n’a aucune valeur. Un petit p à 10^-9 prouve un écart au protocole de randomisation et est donc fortement informatif. Les petits p sont donc utiles s’ils sont bien interprétés. Afin d’éviter une surinterprétation d’un petit p entre 0.01 et 0.05 par un lecteur mal avisé ou un selective reporting d’auteurs qui souhaiteraient dissimuler un écart au protocole, le compromis du NEJM est optimal : fournir les caractéristiques des patients dans chaque groupe, mais ne pas faire de test statistique formel tout en permettant au lecteur avisé de réaliser ces tests a posteriori.s
Argumentaire détaillé
Dans les études observationnelles, on présente ce tableau avec des petits p afin de « montrer » la « comparabilité » des groupes. Je ne m’étendrai pas trop sur les études observationnelles mais traiterai essentiellement du cas des essais cliniques randomisés.
L’argument pour défendre l’absence de présentations des petits p est le fait que, par principe, toute différence observable entre les deux groupes est due au hasard, cela étant garanti par le processus de randomisation. Ainsi, par exemple, une différence d’âge conduisant à un petit p à 0.01 entre les deux groupes, est entièrement due à des fluctuations d’échantillonnage et même si cela désavantage un des deux groupes, cela n’affecte pas l’espérance de la statistique finale. Cela reflète de l’erreur aléatoire d’espérance nulle, pas du biais. Ceux qui veulent réduire cette erreur aléatoire peuvent préalablement spécifier dans le protocole un ajustement sur des facteurs pronostiques majeurs ainsi qu’une stratification de la randomisation sur ces mêmes facteurs (les limites en ont déjà été discutées dans ce blog). En conséquence, le garant de la comparabilité initiale des groupes est le processus de randomisation et aucunement la non-significativité des petits p à baseline. Donc, les petits p ne servent pas à savoir si les groupes sont comparables à baseline.
Il y a encore un autre problème avec la validité statistique des petits p dans le tableau 1. Dès que la randomisation est stratifiée sur un facteur, l’équilibre du facteur entre les deux groupes est garanti comme presque parfait, de telle sorte que les tests usuels (chi², Student), fournissant des petits p uniformes entre 0 et 1 en cas de randomisation simple, auront tendance à fournir des petits p très proches de 1. D’un point de vue strict, les tests employés sont statistiquement erronés.
Ensuite, le problème de multiplicité des tests existe. Souvent il y a 10 ou 20 caractéristiques présentées, et donc 10 ou 20 tests. Il est donc tout à fait attendu d’avoir un ou deux petits p inférieur à 0.05 dans ce tableau, en conditions tout à fait ordinaires, sans la moindre malchance.
Enfin, aussi bien dans les études observationnelles que randomisées, il y a un problème à réaliser un test de différence lorsqu’on souhaite montrer l’équivalence. Ce problème est caricatural dans les études observationnelles de toute petite taille pour lesquelles des différences énormes sont observées dans les groupes et on conclut que les deux groupes ont les mêmes caractéristiques dans les populations dont elles sont tirées puisque les différences peuvent être expliquées par le hasard. Néanmoins, cela ne prouve pas qu’elles SONT expliquées par le hasard. D’un point de vue fréquentiste, seul un test significatif a valeur de preuve. Échouer à montrer une différence ne prouve pas l’équivalence, notamment lorsque la puissance est presque nulle. Si on veut prouver l’équivalence des caractéristiques des groupes, des tests d’équivalence doivent être réalisés plutôt que des tests de différence.
Néanmoins, c’est là que l’interprétation bayésienne doit intervenir. De mon point de vue, le petit p de différence dans un essai clinique randomisé peut être pertinent à condition de placer le seuil de significativité extrêmement bas (10^-6 pour éveiller le soupçon, 10^-9 pour le confirmer). Il ne faut pas négliger le risque d’écart au protocole de randomisation. La qualité de la randomisation fait partie des éléments à prendre en compte dans la lecture critique d’article, et le tableau 1 peut aider à juger de cette qualité. Ainsi, un investigateur d’un centre où il est le seul à inclure peut aisément tricher dans un essai clinique randomisé en ouvert avec une randomisation par blocs de taille 4 stratifiée sur le centre. En effet, l’aléatoire devient très prévisible. Dès que 4 patients ont été randomisés dans le même bras, l’investigateur sait avec certitude que le patient suivant sera randomisé dans l’autre bras. Avec 3 patients randomisés dans le même bras, il a une forte présomption. Cela peut lui permettre de différer ou annuler l’inclusion d’un patient qu’il ne souhaiterait pas voir dans le bras de randomisation certain ou probable. Cela entraînera un déséquilibre entre les groupes sur certaines caractéristiques telle que la sévérité de la maladie. Il peut aussi y avoir un mauvais respect de l’ITT ou une analyse en per protocol qui exclue plus de patients dans un groupe que dans un autre.
C’est ainsi qu’on peut interpréter des petits p comme des détecteurs à défaut de randomisation. Considérons le seuil de significativité 10^-6 et 20 caractéristiques comparées à chaque essai clinique. Si on considère le risque de grosse triche sur la randomisation à une chance sur 100 (je ne sais pas si la réalité est plutôt à 1/10, 1/100 ou 1/1000) et qu’un quart de ces triches soient suffisamment importantes sur des essais cliniques de suffisamment grande taille pour conduire à une différence significative au seuil 10^-6, alors on s’aperçoit qu’un essai sur 400 (1/400=0.0025) sera un essai avec triche de randomisation et p<10^-6 alors que 99/100×(1-(1-10^-6)^20)=1.98×10^-5 seront un essai clinique sans triche de randomisation mais avec un p<10^-6. En présence d’un petit p<10^-6 la probabilité a posteriori de triche est égale à 0.0025/(0.0025+1.98×10^-5) = 99.2%. Si on considère que la grosse triche est seulement probable à une chance sur 1000 et toujours détectable (p<10^-6) une fois sur quatre, alors la probabilité a posteriori est de 92.6%. Le doute est donc permis. Par contre, si on abaisse le seuil de significativité à 10^-9, alors même en considérant que la triche est exceptionnelle (1 chance sur 10 000) et qu’en cas de triche elle ne conduit à une différence significative au seuil 10^-9 qu’une fois sur 100, alors la proba de triche a posteriori d’un résultat significatif à 10^-9 est de 98%.
C’est pourquoi je pense que les tests statistiques restent pertinents, à condition de décaler complètement le seuil de significativité. À 10^-6 un petit p doit éveiller les soupçons et à 10^-9 il y a manifestement un problème, du moins si des tests statistiques robustes ont été appliqués. Par exemple, le test exact de Fisher fonctionne assez bien pour des petits p extrêmes (il a juste tendance à être un peu conservatif) alors que le test du chi² et le test de Student reposent sur un théorème central limite qui est d’autant plus faux qu’on s’intéresse aux queues de probabilité. Il est à noter que ces tests sont plus solides en cas de randomisation 1:1 qui reste le design le plus fréquent.
Il est à noter qu’en cas de stratification, les écarts au protocole sont bien plus faciles à voir. La plupart du temps, seul un déséquilibre minime est possible sur une variable sur laquelle une randomisation stratifiée a été appliquée. Un déséquilibre plus important que la valeur maximale théorique prouve un écart au protocole, sans le moindre doute. L’écart au protocole peut être modeste (p.e. usage d’un nombre de listes de randomisation plus élevé que précisé dans le protocole, ou usage de blocs de plus grande taille) ou pas.
Faut-il rapporter les petits p dans les tableaux 1 des essais cliniques randomisés ? Faut-il rapporter seulement les caractéristiques de tous les groupes poolés sans test ? Faut-il séparer chaque groupe sans les comparer statistiquement (comme dans le NEJM) ?
Mon opinion personnelle, c’est que l’approche du NEJM est optimale. Elle permet d’éviter les surinterprétations de petits p entre 0.01 et 0.05 de lecteurs mal avisés mais permet aussi au lecteur avisé de calculer a posteriori un petit p à 10^-9 s’il a un soupçon. Enfin, elle permet l’identification d’écarts au protocole sur la stratification de la randomisation, qui ne nécessite pas de calcul de petit p mais juste une comparaison brute de l’écart d’équilibre entre les groupes par rapport au déséquilibre théorique maximal. Enfin, un bénéfice non négligeable, à mon avis, c’est la réduction du risque de selective reporting. Si le petit p doit être calculé par les auteurs, ceux-ci sont susceptibles de supprimer la caractéristique considérée du tableau 1, par crainte que cela bloque la publication (selective reporting bias). S’ils ne testent pas, cela peut être identifié lors du reviewing (avec ou sans refus de l’article) ou lors de la lecture critique de l’article publié ; deux options meilleures que la dissimulation.
Certains statisticiens orientent le choix des analyses statistiques par les résultats d’autres statistiques. Un grand classique, est illustré dans une « comparaison de deux groupes » sur une variable quantitative. Un test de normalité (p.e. Shapiro-Wilk) est d’abord fait. Si le test est significatif, alors la distribution est considérée comme non-normale et un test de Mann-Whitney est réalisé, autrement un test de Student sur séries indépendantes est réalisé. Éventuellement, un test d’égalité des variances (test de Levene ou de Bartlett) est réalisé pour décider de l’usage d’une variante de Student sans hypothèse d’égalité des variances (Aspin-Welch).
Ne faites pas ça ! Cela va à l’encontre d’un grand nombre de principes statistiques fréquentistes :
La question de recherche décide de la statistique à comparer
Un même protocole doit conduire à une même statistique
La validité de chacun de ses tests repose sur une analyse des fluctuations d’échantillonnage qui n’est valide que lorsque la statistique est reproduite à l’identique à tous les coups
La question de recherche décide de la statistique à comparer
D’abord, le test de Mann-Whitney n’est pas un test de comparaison de moyennes. Si l’objet de l’étude est de comparer les coûts moyens de prise en charge, par exemple, on peut craindre que le test fasse une erreur de 3ème espèce, c’est-à-dire conclure à un coût moyen plus bas dans le groupe où il est plus élevé.
Le code R suivant montre un exemple dans lequel les conclusions des deux tests sont opposées :
Ce phénomène paradoxal s’applique notamment à une intervention ayant un coût fixe non négligeable mais évitant des surcoûts rares et élevés. On peut, par exemple, imaginer qu’une mesure de prévention telle que l’antibioprophylaxie pour certaines chirurgies, coûte quelques euros à chaque intervention (p.e. 30 €) mais économise des milliers d’euros (p.e. 3000 € en moyenne) pour un sujet sur 50. Dans ces conditions, le coût moyen de prise en charge est plus bas avec l’intervention alors que le test de Mann-Whitney conclura à un coût plus élevé !
Cela est dû au fait que le test de Mann-Whitney ne compare pas les moyennes mais compare l’aire sous la courbe ROC à 0.50, en considérant que le test est la variable quantitative (p.e. durée de séjour) et le groupe est le diagnostic binaire. On notera au passage que le test de Mann-Whitney ne compare pas les médianes. Il m’est déjà arrivé d’observer des durées médianes de séjour identiques entre deux groupes alors que le test de Mann-Whitney montrait une différence significative.
Si on s’intéresse au coût moyen, alors on fera un test de comparaison de moyennes. Si on s’intéresse à une capacité diagnostique d’un dosage biologique, par exemple, alors on tracera une courbe ROC et le test de Mann-Whitney aura un certain sens (même si la comparaison à 0.50 n’est pas forcément très pertinente).
Un même protocole doit conduire à une même statistique
Avec la méthode de Student, on peut fournir un intervalle de confiance à 95% de la différence de moyennes. Si le protocole était répété de très nombreuses fois, dans 95% des cas, l’intervalle de confiance contiendrait la « vraie » différence de moyennes dans la population. C’est ce qu’on appelle la couverture de l’intervalle de confiance. Le test de Shapiro-Wilk étant aléatoire, de manière aléatoire, un Mann-Whitney ou un Student sera réalisé. Si la puissance du Shapiro-Wilk est de 50%, alors un Student sera fait une fois sur deux. Comment définir alors la couverture ? On peut définir la couverture conditionnelle au fait que la statistique soit générée. Auquel cas, on s’aperçoit que la statistique de Shapiro-Wilk n’est pas indépendant du résultat du test de Student, et conduit à un biais de couverture très important !
Analyse des fluctuations d’échantillonnage conditionnelles
set.seed(2020)
delta=2
v=t(sapply(1:100000, function (x) {
a=rexp(30)
b=delta+rexp(30)
if (shapiro.test(c(a,b))$p.value<0.05) {
return (c(NA,NA))
} else {
tt = t.test(b,a,var.equal=T)
return(tt$conf.int)
}
}))
mean(is.na(v[,1])) # puissance du test à 95%
# on conditionne à un Shapiro non significatif
w=v[!is.na(v[,1]),]
# On calcule les défauts de couverture
# risque borne haute 12.7% (théorie 2.5%)
mean(w[,2] < delta)
#risque borne basse 0% (théorie 2.5%)
mean(w[,1] > delta)
Ce code R montre qu’avec deux échantillons de taille 30 suivant deux lois exponentielles décalées de 2, l’intervalle de confiance à 95%, au lieu d’avoir un risque de 2.5% de surestimation et 2.5% de sous-estimation, aura un risque de 12.7% de sous-estimation et de 0% de surestimation. Cela est un biais majeur ! Si on enlève le test de Shapiro-Wilk, les risques passent à 2.4% de chaque, très proche du risque nominal de 2.5% de chaque côté. En bref, le test de Student est robuste aux écarts à la normalité, mais la séquence Shapiro->Student est complètement biaisé.
Analyse des fluctuations du petit p
Enfin, considérons les fluctuations d’échantillonnage d’un petit p issu aléatoirement d’un test de Mann-Whitney ou de Student selon le résultat d’un Shapiro-Wilk. Il est possible que les moyennes des deux groupes soient égales (première hypothèse nulle) mais que le test soit presque toujours significatif (car le test de Shapiro-Wilk est suffisamment puissant et les moyennes de rang sont inégales) conduisant à un risque alpha tendant vers 100% comme dans l’exemple ci-dessous, sous le logiciel R :
# moyennes égales...
# mais moyennes des rangs différentes
set.seed(2020)
v=sapply(1:1000, function (x) {
a=rexp(2500) # moyenne=1
b=rnorm(2500)+1 # moyenne=1
if (shapiro.test(c(a,b))$p.value<0.05) {
wilcox.test(a,b)$p.value
} else {
t.test(a,b,var.equal=T)$p.value
}
})
mean(v<0.05) # Risque alpha 99.8%
Sous la seconde hypothèse nulle, d’égalité des moyennes des rangs (hypothèse nulle de Mann-Whitney), alors il est possible d’avoir un risque alpha élevé aussi si le test de Student est souvent réalisé.
Sous l’hypothèse nulle de distribution parfaitement superposées, alors le problème est nettement moindre. Néanmoins cette hypothèse nulle peut être testée beaucoup plus efficacement par le test de Kolmogorov-Smirnov mais elle n’a aucune pertinence. Par exemple, un groupe peut simplement avoir une variance différente de l’autre, sans pour autant que la moyenne, la médiane ou la moyenne des rangs ne diffère. Un test prouvant que les deux distributions ne sont pas superposées ne permet nullement de savoir laquelle des deux distributions est la « meilleure » dès lors qu’on s’intéresse à une exposition potentiellement bénéfique (p.e. traitement) ou nocive (p.e. exposition épidémiologique).
Pour les méta-analyses
Un mélange de statistiques est presque inexploitable. Les méta-analystes sont obligés de bidouiller. Souvent ils vont approximer la médiane à la moyenne si cette première est fournie.
Au final
Le test de Student est assez robuste aux écarts à la normalité. Le test de Mann-Whitney n’est pas un test de comparaison de moyennes. La séquence des tests est une ineptie statistique. Ce problème existe aussi avec d’autres séquences de tests, comme on en retrouve, par exemple, dans les méthodes pas-à-pas, les comparaisons de modèles guidant la suite des analyses (p.e. comparaisons de plusieurs polynômes fractionnaires, tests de linéarité, comparaisons de transformations, tests « omnibus » avant de réaliser des comparaisons deux à deux, etc.)
Le choix d’une statistique doit être guidé par des considérations théoriques. Lorsqu’on s’intéresse à une différence des moyennes, plusieurs estimateurs sont possibles (Student, bootstrap) et encore une fois, ce choix doit se faire a priori plutôt qu’aléatoirement parce que chacune des procédures n’est valide que lorsqu’elle est réalisée inconditionnellement.
Voici quelques exemples d’augmentation du risque alpha (encore appelé chance alpha) du fait de P-hacking sur les ajustements. L’analyse statistique est réalisée dans un modèle linéaire général avec petit p calculé selon une loi de Student à partir de la variance des coefficients estimée sur l’échantillon (méthode de Wald).
Le scénario consiste en une randomisation simple 1:1 de N=100 sujets (~50 par groupe) avec un outcome quantitative suivant une loi normale et 4 covariables suivant aussi des lois normales.
Le P-hacking est basé sur les libertés suivantes :
Ne pas mettre la covariable du tout dans le modèle
Mettre la covariable en effet quantitatif linéaire
Mettre la covariable dichotomisée sur la médiane
Découper la variable en tertiles, quartiles ou quintiles
Et ce, séparément pour chaque variable
D’autres recodages auraient pu être envisagés, tels que le découpage à des seuils « conventionnels » (p.e. tranches de 10 ou 15 ans pour l’âge) ou l’usage de polynômes fractionnaires. Je suis resté simple. L’usage de trop de techniques n’est pas non plus habituel dans les essais cliniques randomisés.
Le P-hacking est obtenu par l’exécution de toutes les combinaisons possibles et la sélection du petit p minimal.
Dans le premier scenario, les covariables sont de réelles covariables pronostiques avec des effets beta=+1 (+1 écart-type d’outcome par écart-type de variable), beta=+0.50, beta=+0.25 et beta=+0.10 pour les 4 covariables, c’est-à-dire une covariable fortement pronostique, une moyennement pronostique et deux faiblement pronostiques. Pour un seuil de significativité bilatéral à 5%, dans ce scenario le risque (ou chance) alpha bilatéral monte à 24.7% (incertitude 24.2 à 25.2%). NB: cela correspond à une chance alpha unilatérale deux fois plus petite. C’est cette chance unilatérale qui intéresse généralement le P-hackeur.
Le second scenario est identique au premier à ceci près que le non-ajustement sur une covariable est interdit. Cela correspondait au respect d’un protocole précisant la liste des covariables d’ajustement sans en préciser le codage. La chance alpha bilatérale est à 20.9% (incertitude 20.5 à 21.4%).
Dans le troisième scenario, le non-ajustement sur une covariable est toujours interdit, mais en plus aucune des covariables n’est corrélée à l’outcome (les 4 effets sont nuls). La chance alpha redescend à 8.1% (incertitude 7.8 à 8.4%).
Le quatrième scenario est identique au troisième à ceci près que 200 sujets sont randomisés plutôt que 100. La chance alpha descend à 7.1% (incertitude 6.8 à 7.4%)
Le cinquième scenario est identique au second à ceci près que la taille d’échantillon est de 400 plutôt que 100. La chance alpha passe de 20.9% (pour N=100) à 19.8% (pour N=400, incertitude 19.4 à 20.2%).
Le sixième scenario est assez différent. Il est basé sur 20 covariables non corrélées à l’outcome. Ainsi, on compense l’absence de facteur pronostique par le nombre de variables testées. L’algorithme de P-hacking est très allégé car le nombre de combinaisons possibles est trop grand. On se contente alors de choisir en analyse trivariée (outcome ~ traitement+covariable) pour chaque covariable, le codage optimal (améliorant le petit p dans le sens de la supériorité du traitement innovant), y compris la suppression de la covariable. Une fois chacune des covariables analysée, on intègre toutes ces covariables codées comme il faut, dans le modèle le « meilleur ». Ce sixième scenario est réalisé avec N=100 et la liberté de supprimer des covariables, comme vous l’aurez compris. La chance alpha bilatérale montait à 37.4% (incertitude 36.6% à 38.2%).
Ainsi, il semblerait que le P-hacking soit efficace, même lorsque les covariables d’ajustement sont précisées dans le protocole (mais pas leur codage), mais son efficacité dépend beaucoup du degré de corrélation entre les variables pronostiques et l’outcome. Si elles sont fortement corrélées à l’outcome, alors les corrélations négatives qui peuvent aléatoirement apparaître entre le traitement innovant et le facteur pronostique vont booster le petit p d’une manière dépendante de la force pronostique. Cette force pronostique est la somme d’une partie fixe (pour les vrais facteurs pronostiques) et d’une partie aléatoire d’autant plus grande que l’échantillon est petit. C’est pourquoi, si on veut maximiser sa chance alpha, il faut rechercher de vrais facteurs pronostiques lorsque l’échantillon est grand. Pour les petits échantillons, on peut se concentrer sur de multiples variables peu ou pas corrélées à l’outcome.
Je n’ai pas analysé la chance alpha induite par l’imputation des données manquantes. Cela n’est pas évident de sélectionner des scenarii plausibles.
Il serait possible de réaliser ce travail de simulations avec une étude réelle, de telle sorte que les facteurs pronostiques aient des corrélations « de la vraie vie » avec l’outcome.
Billet sur un sujet d’actualité : le virus SARS-CoV-2 responsable de l’épidémie de COVID-19. Le Pr Didier Raoult, virologue français, fait la promotion du traitement par hydroxychloroquine pour le COVID-19. Notre ministre de la santé a annoncé qu’un essai clinique plus vaste soit initié, ce qui me paraît raisonnable. Que peut-on dire de la qualité méthodologique et de la fiabilité des conclusions de l’essai clinique non randomisé Marseillais ?
Si je dois faire bref : il s’agit d’un essai clinique de petite taille, de méthodologie très médiocre, plutôt mal conduit, comportant de nombreux biais et dont les conclusions doivent être pris avec des pincettes. La confirmation dans un essai clinique randomisé de grande taille, avec un critère de jugement clinique, me paraît indispensable.
Critère de jugement
Le critère de jugement, purement biologique (négativation de la PCR virale) est pertinent pour un essai clinique de petite taille mais sa corrélation à un meilleur résultat clinique est loin d’être sûre, d’autant qu’il y a trois passages en réanimation, un décès et une interruption de traitement pour effet indésirable sur 26 patients traités par hydroxychloroquine, soit 5/26 = 19% (IC95% : 6.6% à 39.4%) de résultats cliniquement non satisfaisants.
Il est à noter que le critère de jugement principal décrit dans l’article est la négativation de la PCR virale à J6 alors que le protocole EudraCT parle de détection virale à J1, J4, J7 et J14, sans hiérarchiser. En bref, cela suppose des tests multiples et une correction de multiplicité des tests, et en aucun cas une détection virale à J6.
Critères d’inclusion et biais de sélection
L’essai clinique n’est pas randomisé. Le groupe intervention est constitué de patients de Marseille ayant accepté la participation à l’essai clinique à Marseille et respectant les critères d’inclusion : patient hospitalisé d’âge supérieur ou égal à 12 ans et PCR SARS-CoV-2 sur échantillon naso-pharyngé positif à l’admission, quel que soit l’état clinique. Les femmes enceintes et les sujets ayant des contre-indications à l’hydroxychloroquine étaient exclus.
Jusque là, des critères plutôt classiques, assez larges mais pragmatiques (population d’inclusion proche de la population susceptible de bénéficer du traitement), permettant une inclusion rapide de patients.
L’échantillon contrôle est beaucoup plus douteux. C’est un mélange de patients ayant de Marseille refusé l’hydroxychloroquine (nombre non précisé) et de patients recrutés à Nice, Briançon et Avignon; centres dans lesquels aucun patient ne bénéficiait du traitement innovant. Il semble étonnant que le traitement ne soit pas proposé dans les autres centres et on peut craindre que les patients des autres centres aient été recrutés de manière assez chaotique, en dehors du cadre de la recherche. Que dit le protocole posté sur EudraCT. La section E.8 est assez claire : l’essai n’est pas contrôlé (E.8.1) et il est monocentrique (E.8.3). Le protocole prévoyait 25 sujets, ce qui est cohérent avec les 26 recrutés dans le bras expérimental de l’étude (20 analysés).
Les doutes quant à la qualité du groupe contrôle sont confirmés par les données brutes fournies en annexe. Elles montrent 2 données manquantes sur le dosage de la charge virale à l’admission (J0) dans le groupe contrôle. Ces 2 sujets bénéficient d’une première PCR à J2 et à J3 respectivement. Comment est-il possible de ne pas rechercher le virus à J0 alors que ça fait partie des critères d’inclusion ? On peut craindre que les patients aient été inclus dans l’étude a posteriori, après qu’une charge virale ait été mesurée à J2 ou J3 alors que le dosage n’avait pas été fait initialement. Ce problème aurait été évité si l’étude avait bénéficié d’un e-CRF centralisé avec inclusion des patients au fur et à mesure. On peut craindre un biais de sélection secondaire important du groupe contrôle. Durant le temps où Marseille est arrivé à inclure 26 patients, les trois autres centres sont arrivés, au maximum à recruter 16 patients, sans compter la partie recrutée à Marseille. On peut donc craindre que plus de patients que ça étaient éligibles mais que seuls 16 ont été sélectionnés selon des critères que nous ne connaissons pas.
Les données publiées permettent aussi de constater que deux patients du groupe contrôle ont 10 ans et donc, ne respectent pas les critères d’inclusion (âge >= 12 ans). On constate aussi sur ces données que le statut virologique n’est pas renseigné de la même manière chez tous les patients. Pour 6 patients contrôles sur 16, le nombre de cycles (CT) de PCR est précisé lorsqu’il est en dessous de 35 alors que pour les 10 autres, la donnée est binaire « positif » ou « négatif ». Sur les 20 patients du groupe hydroxychloroquine, tous ont des mesures quantitatives. Ce défaut de standardisation des données suggèrerait l’usage d’un tableur Excel (ou LibreOffice) plutôt qu’un e-CRF pour la gestion des données, avec le défaut de traçabilité et le risque de biais de sélection secondaire qu’on peut imaginer.
Exclusions
Sur les 26 patients ayant bénéficié de l’hydroxychloroquine, 6 ont été exclus des analyses. Cela a déjà été mentionné plus haut, mais exclure un sujet décédé, trois passages en réanimation, une interruption de traitement pour effet indésirable, pour évaluer la « guérison virale » est méthodologiquement inacceptable. Le seul patient pour lequel la question se pose, c’est le sujet sorti d’hospitalisation qui avait probablement un bon état clinique. Une analyse plus pertinente aurait été une analyse en intention de traiter, en considérant tout passage en réa ou décès comme un échec. Les autres patients, notamment l’interruption thérapeutique auraient dus être suivi virologiquement comme les autres et jugés comme les autres.
Il est à noter que le résultat de l’analyse principale reste significative même en considérant que tous ces patients sont des échecs (équivalent à une positivité virale à J6). Ainsi, la mauvaise méthodologie ne remet pas forcément en cause complètement les résultats.
Données manquantes
Il y a BEAUCOUP de données manquantes et leur fréquence diffère beaucoup selon le groupe : 7/140 (5%) dans le groupe hydroxychloroquine contre 43/112 (38.4%) dans le groupe contrôle, ce qui confirme encore la non-comparabilité des groupes. Par ailleurs, ces données manquantes sont aussi présentes au moment de l’évaluation du critère de jugement principal à J6 : 1/20 (5%) dans le groupe hydroxychloroquine vs 5/16 (31.2%) dans le groupe contrôle. Une imputation LOCF a été effectuée. Même si cette imputation n’est pas aberrante, cela affecte de manière non négligeable la précision statistique (surestimée avec l’imputation simple). Par ailleurs, cela désavantage le groupe avec le plus de données manquantes (groupe contrôle) car moins de temps est laissé au patient pour se négativer. Par exemple le patient 15 (groupe contrôle) a été inclus sur un dosage virologique à J3 (écart au protocole) et a seulement eu le droit à un second dosage à J5. Par principe, il ne lui a été donné que deux jours pour se négativer là où les sujets du groupe témoin avaient généralement 6 jours.
On remarquera aussi que les détection virales « clignotent ». Certains sujets positifs, se négativent à un moment puis se re-positivent plus tard. Ainsi, 4/16 sujets clignotent dans le groupe contrôle et 4/20 sujets clignotent dans le groupe hydroxychloroquine. Cela fait douter de la qualité des prélèvements ou de la fiabilité de la PCR. Cela pose aussi le problème de la fiabilité de l’imputation LOCF puisque le sujet du groupe hydroxychloroquine, négatif aux dernières nouvelles, pour lequel on n’a pas recherché le virus à J6 est avantagé par cette imputation (étant donné qu’il aurait pu se re-positiver) tandis que les 5 sujets du groupe contrôle ont été désavantagés par la non-mesure de la charge virale à J6 puisqu’ils étaient positifs aux dernières nouvelles.
On peut encore remarquer que le groupe contrôle se divise en deux : les sujets avec quantification (n=6) et les sujets sans quantification (n=10). Il est possible que ces sujets avec quantification corresponde aux sujets de Marseille (ce ne serait pas étonnant, vu qu’il y a 4 enfants asymptomatiques sur six patients, pour lesquels on peut comprendre que les parents aient refusé la participation à un essai clinique sur un traitement innovant alors que tout allait bien pour leur petit bout de chou) ou alors il s’agit au moins de sujets suivis avec une « quantification précise » de la PCR. Considérons donc le groupe contrôle. Sur les 6 sujets avec quantification précise, 5 ont au moins une virologie négative à un moment où à un autre contre 0 sur les 10 sujets sans quantification précise (p=0.0014 selon un test exact de Fisher). Comme il s’agit d’une analyse post hoc, ce résultat est à prendre avec des pincettes (au même sens que l’analyse très douteuse sur l’Azithromycine). Une partie de la différence est explicable par le nombre de données manquantes extrêmement grand chez les sujets sans suivi rigoureux (38/70=54%) baissant les chances d’avoir un prélèvement négatif dans le long. Néanmoins, on peut douter de la comparabilité des prélèvements des patients sans quantification précise : le nombre de cycles de PCR était-il le même ? Les conditions de prélèvement étaient-elles les mêmes ?
Réanalyse avec nouvelle gestion des données manquantes
D’abord, les six sujets du groupe hydroxychloroquine exclus ont été réintroduits avec l’imputation suivante : le décédé et les sujets passés en réa ont été considérés comme d’évolution favorable (positif à J6). Les deux autres sujets (effet indésirable et sortie d’hospitalisation) ont été analysés sur la base des données disponibles, les prélèvements ultérieurs étant considérés comme « non faits ».
La première ré-analyse a été faite avec une imputation multiple par équations chaînées selon la méthode du package mice. Les variables utilisées pour l’imputation étaient : le groupe de traitement et chacun des dosages binaires (positif ou négatif) de D0 à D6. Chaque variable était imputée dans un modèle de régression logistique expliqué par toutes les autres variables. Un total de 500 jeux d’imputation a été réalisé. Une régression logistique bivariée expliquant la positivité à J6 selon le groupe de traitmeent était estimé par le maximum de vraisemblance pour chaque jeu d’imputation. Les log-odds-ratio ont été poolés avec la méthode de Rubin, c’est-à-dire, globalement en additionnant la variance intra à la variance inter. La méthode de Wald, prenant en compte le nombre de degrés de liberté a été finalement utilisé pour estimer le degré de significativité de la statistique poolée ainsi que son intervalle de confiance.
La fraction d’information manquante (FMI) était estimée à 0.26. L’odds ratio de l’effet du traitement était estimé à 0.143 (IC95% : 0.023 à 0.884, p=0.037). Cette estimation est très fragile car les conditions de validité asymptotiques des méthodes utilisées ne sont pas atteintes.
Une autre méthode de calcul, c’est simplement un test exact de Fisher en excluant les sujets avec une évaluation manquante à J6. On obtient alors un odds ratio à 0.18 (IC95% : 0.016 à 1.15, p=0.064).
Il semble donc que la tendance statistique persiste avec ces analyses de sensibilité. Néanmoins, la fiabilité de résultats en présence d’une mauvaise qualité de données reste toujours sujette à caution.
Deuxième ré-analyse : comparaison à des données historiques
Problème de multiplicité des tests
La comparaison entre les groupes est répétée tous les jours de J1 à J6, soit six tests pour la comparaison hydroxychloroquine vs contrôle. Cela peut paraître beaucoup mais ne me préoccupe pas tant que ça. D’abord, l’analyse principale est annoncée comme la comparaison à J6. C’est néanmoins douteux car ça ne correspond pas au protocole EudraCT. On peut alors craindre un choix a posteriori du critère de jugement principal et donc craindre du P-hacking. Sans préjuger d’autres formes de P-hacking, la sémiologie du P-hacking par multiplicité des tests n’est pas retrouvée. Cette sémiologie correspond à un résultat à la limite de la significativité statistique (p entre 0.01 et 0.05), sans cohérence globale telle qu’une différence qui aurait un p=0.03 à J4 mais un p=0.30 à J3 et un p=0.40 à J5. Dans cette étude, les différences entre les deux groupes sont retrouvées de manière cohérente avec un écart progressivement croissant et une forte significativité. Le bémol, c’est que les écarts se resserrent dans les analyses de sensibilité que j’ai réalisées. C’est donc moins robuste qu’il paraît.
Par contre, l’analyse en sous-groupe sur l’Azithromycine+Hydroxychloroquine est très douteuse. Elle n’était absolument pas planifiée dans le protocole EudraCT et pourtant se retrouve dans le titre de l’article, correspond à un sous-groupe minuscule (n=6). Déjà, le test statistique employé est incorrect. C’est un test global sur les trois groupes, répondant à la question : y a-t-il un au moins des trois groupes dont le pourcentage de négativation à J6 diffère des autres ? Pour conclure au bénéfice de l’Azithromycine en addition à l’Hydroxychloroquine et en considérant que cette dernière a déjà fait preuve de son efficacité (avec tous les bémols mentionnés au dessus), alors il faudrait comparer le groupe Hydroxychloroquine seul à Azithromycine+Hydroxychloroquine. Avec le test exact de Fisher, le taux de succès à J6 de 8/14 n’est pas significativement différent de 6/6 (p=0.115). Par ailleurs, d’autres analyses en sous-groupes étaient possibles : par exemple Azithromycine seule vs contrôle ou Azithromycine±Hydroxychloroquine vs Autre. Il aurait pu aussi y avoir d’autres analyses en sous-groupes sur la prise en charge… On ne peut pas savoir combien ont été faites étant donné qu’aucun protocole publié ne fournit d’information dessus.
D’une manière générale, on évite les analyses post hoc en sous-groupes dans les essais cliniques, mais c’est encore plus vrai lorsqu’ils sont de toute petite taille, car la puissance est proche du risque alpha, conduisant à un risque de fausse découverte très élevé.
Ajustements statistiques ?
Les effectifs sont tellement petits qu’il est difficile de faire une comparaison statistique des caractéristiques des patients. Ces comparaisons ont tendance à être non significatives alors que des différences majeures existent entre les groupes. Ces différences peuvent être expliquées par les fluctuations d’échantillonnage, ou être expliquées par une sélection différente des patients, mais faire la part des choses est très difficile. On a quand même quelques indices pour penser que la population traitée a une sévérité de la maladie un plus élevée : trois passages en réanimation et un décès sur 26 patients (versus zéro sur 16 ?). Une moyenne d’âge plus élevée (52.1±18.7 vs 37.3±24 ans, p=0.06) chez les sujets non exclus. La différence aurait peut-être été encore plus forte si on avait inclus les patients passés en réa et décédés puisque ces risques touchent quand même nettement plus les personnes âgées. Archétype de la variable impossible à comparer : la proportion d’infections respiratoires basses (LRTI) est de 30% dans le groupe hydroxychloroquine vs 12.5% dans le groupe contrôle, mais le nombre de sujets est tellement bas que ça peut être complètement expliqué par le hasard (p=0.30).
Certains pourraient proposer des ajustements statistiques. C’est difficile avec des effectifs aussi faibles. On peut juste se demander dans quel sens va le biais. La charge virale semble plutôt positivement corrélée à la sévérité (https://doi.org/10.1016/S1473-3099(20)30232-2) et la clairance semble être plus lente dans les formes sévères. Ainsi, le groupe hydroxychloroquine est a priori désavantagé par ce biais de sélection différentiel des formes plutôt sévères. Cela ne remet donc pas en cause les résultats.
Nombre de sujets nécessaires
L’article comporte un calcul de nombre de sujets nécessaires comme si l’étude avait été planifiée avec un groupe contrôle dès le départ. Cela semble être un calcul a posteriori. Je n’ai pas de preuve absolue mais le fait que le protocole EudraCT soit clairement conçu comme un essai à un seul bras et est un premier indice. Le second indice est le fait que le groupe contrôle soit construit d’une manière très étrange, mélangeant des refus de participation à l’étude (j’espère qu’ils ont quand même signé un consentement pour avoir des PCR tous les jours, sinon le CPP pourrait ne pas approuver) et des patients des centres non Marseillais pour lesquels le traitement n’était pas une option. L’inclusion de ce dernier groupe de patients non Marseillais paraîtrait éthiquement douteux si les patients avaient des prélèvements sanguins de PCR spécifiquement pour la recherche sans avoir la possibilité de bénéficier du traitement innovant ; mais il paraît possible qu’ils aient simplement été inclus comme une série de cas rétrospective à partir des dossiers médicaux et qu’en conséquence ils n’aient eu aucun examen ou traitement en dehors de la routine.
Au total
Le biais le plus préoccupant de cette étude, à mon avis, provient du suivi et surtout de la sélection des contrôles en dehors de Marseille. Certains de ces sujets n’ont pas eu de PCR à baseline mais ont quand même été inclus. De très nombreuses données manquent sur leur PCR virales. Leur PCR est considérée comme « positive » ou « négative » sans quantification, pour la plupart. On peut se permettre de douter du fait que les prélèvements ont été réalisés, cultivés et analysés de la même manière qu’à Marseille. Mais le pire, c’est qu’on ne sait pas quels sujets étaient éligibles dans le groupe contrôle mais n’ont pas été inclus dans l’étude.
Sans ce problème de groupe contrôle, le résultat principal à J6 aurait une certaine robustesse malgré les données manquantes et la multiplicité potentielle des tests (puisque le CJP n’est pas celui du protocole). Du fait de ce groupe contrôle douteux, j’émets des réserves quand à la reproductibilité de l’analyse principale.
Cela ne veut pas dire que le traitement est inefficace ou que l’article n’apporte rien. Le résultat est très fragile mais fournit quand même une piste intéressante, à mon avis. Je me range du côté de la communauté scientifique qui propose de réaliser un essai clinique randomisé de grande ampleur afin de statuer sur l’efficacité clinique de l’hydroxychloroquine avec une méthodologie robuste.
Ajout post hoc : comparaison à des données historiques
Comme décrit avant, la fragilité principale de l’étude provient de la sélection du groupe contrôle sans dosage quantitatif précis (c’est-à-dire avec juste POSITIF ou NEGATIF comme résultat de la PCR). On peut craindre qu’il s’agisse de données de routine rétrospectivement collectées, avec un biais de sélection. On peut notamment craindre qu’il y ait eu l’exigence de la présence d’un prélèvement à J5 ou J6 pour inclure le patient. Cela risque d’exclure les patients ayant eu une négativation précoce pour lesquels il n’y a plus de raison de continuer les prélèvements au delà d’un ou deux résultats négatifs.
C’est pourquoi il est encore préférable de comparer l’échantillon hydroxychloroquine à un échantillon indépendant, de données historiques, dont la rigueur de recueil est meilleure. Prenons comme référence l’article Viral dynamics in mild and severe cases of COVID-19. L’article ne présente que les différences de Ct par rapport au prélèvement initial (delta(Ct)). On peut néanmoins comprendre, de la figure, que les sujets n’ont plus de prélèvements une fois négatifs. Dans les formes légères la majorité des sujets est négatif à J8. À J6, il y a nettement plus de positifs : environ la moitié des patients ayant une forme légères. Les formes sévères (dont 23/30=77% sont passés en unité de soins intensifs) par contre, nécessitent nettement plus de temps (> 15 jours pour la négativation). Si on considère l’échantillon des 20 patients dont les cas réanimatoires a été exclu, la majorité doit ne pas être sévère. Un taux de 50% de négativation à J6 paraît plausible. Le taux observé est de 14/20=70% (IC95% : 46 à 88%), compatible avec le taux de 50% ; mais il y a une tendance statistique à un taux plus élevé.
La conclusion reste la même. Un effet biologique paraît plausible mais il y a un doute non négligeable. Le bénéfice clinique reste très douteux. Avant d’arroser la population d’hydroxychloroquine, je conseillerai d’inclure des patients dans l’essai clinique de grande taille ou d’en attendre le résultat.
Ce billet a pour objet de s’adresser à un public statistique moins aguerri que d’autres. Vous avez peut-être déjà entendu parler d’effet marginal ou conditionnel ou encore de modèle marginal ou conditionnel. Qu’est-ce que cela veut dire ou implique ?
Cette problématique s’applique aux régressions logistiques, aux régressions de Poisson, mais pas au modèle linéaire général. En bref, le problème se pose à partir du moment où la relation entre les variables explicatives et la variable à expliquer n’est pas linéaire. Cela s’illustre très bien avec un exemple. Considérons une population dans laquelle un risque (par exemple le décès à 30 jours) va toucher 52.5% de la population. Il existe un facteur pronostique majeur binaire, de prévalence 50%, divisant la population en deux sous-populations de taille égale : une sous-population à bas risque avec 10% de décès et une population à haut risque avec 95%. Comme les deux sous-populations représentent chacune 50% de la population globale, le risque général est bien de 0.50×0.10+0.50×0.95=52.5%.
Supposons qu’un traitement réduise le risque de 5% dans chacune des deux sous-populations. La sous-population à bas risque passe d’un risque de 10% à 5% et le haut risque de 95% passe à 90%. L’odds ratio protecteur du traitement, dans chacune des deux sous-populations est environ égal à 0.50. Pour être rigoureux, il est égal à 0.474 dans chacune des deux sous-populations. C’est l’odds ratio conditionnel au facteur pronostique.
Maintenant, on peut calculer que le risque général d’une population totalement exposée au traitement. La réduction de 5% du risque dans chacun des deux sous-groupes se traduit par une réduction de 5% du risque général qui passe de 52.5% à 47.5%. Cela peut aussi se calculer comme 0.50×0.05+0.50×0.90=47.5%. Ainsi, en calculant 52.5%-47.5% on retombe sur les 5% de réduction. La moyenne des différences (-5% dans chaque sous-population) est égale à la différence des moyennes (52.5% – 47.5%).
Grace à ce second calcul, nous pouvons calculer l’odds ratio protecteur du traitement sur la population générale. Le risque de 52.5% correspond à une cote de 0.525/(1-0.525)=1.105, qui est réduite à un risque de 47.5% soit une cote de 0.475/(1-0.475)=0.905. Au total, l’odds ratio est de 0.905/1.105 = 0.819. Cet odds ratio s’applique au pourcentage moyen de la population complète. C’est un odds ratio de la moyenne, ou odds ratio marginal.
Ainsi, nous avons noté que l’odds ratio de 0.819 est un odds ratio marginal alors que l’odds ratio de 0.475 est un odds ratio conditionnel au facteur pronostique, c’est-à-dire, s’appliquant à chacun des sous-groupes défini par ce facteur pronostique.
Que se passe-t-il lorsque les effets dans chacun des deux sous-groupes ne sont pas égaux ? Certains modèles, comme la régression logistique, reposent sur l’hypothèse de constance des effets conditionnels dans les sous-groupes. En cas d’écart à cette condition, en pratique, l’odds ratio calculé sera égal à une valeur intermédiaire, une sorte de moyenne de tous les odds ratio des sous-groupes, pondérée par la précision statistique de l’odds ratio dans ce sous-groupe. En bref, on peut à peu près obtenir cet odds ratio conditionnel « moyen » en calculant la moyenne des log-odds ratio pondérée par l’inverse de leur variance, puis en repassant à l’odds ratio par l’exponentielle. Selon la manière d’estimer le modèle, on obtiendra un résultat légèrement différent. Point important, il est à noter que les procédures de régression logistique implémentés par tous les logiciels fournissent toujours, en sortie, des odds ratio de chacune des covariable, conditionnels à l’ensemble des autres covariables.
On peut dire que l’odds ratio conditionnel s’applique séparément à chacune des observations, avec son risque de base propre (10% ou 95%), alors que l’odds ratio marginal est l’odds ratio qui s’applique à la moyenne de la population (risque de base 52.5%).
Quel est le problème des odds ratio conditionnels ? D’abord, le terme est incomplet… Conditionnel à quoi ? Un odds ratio conditionnel à l’âge, n’est pas le même qu’un odds ratio conditionnel au sexe, qui diffère encore de l’odds ratio conditionnel à l’âge et au sexe. Ensuite, plus le nombre de conditions est élevé et les facteurs pronostiques fortement liés à l’outcome, plus l’odds ratio conditionnel tend vers 0 ou l’infini. Dans l’exemple, l’odds ratio marginal était faible (0.819) alors que l’odds ratio conditionnel était nettement plus fort (0.475). Cela est explicable par le fait qu’une différence de 5% correspond à un odds ratio faible lorsqu’on est proche d’un risque de 50% (population générale) mais correspond à un odds ratio fort dès que le risque se rapproche de 0% ou 100%. Or, plus un modèle sera fortement prédictif, plus les sous-groupes auront des valeurs prédites extrêmes (proches de 0 et 100%) et plus les odds ratio conditionnels seront forts. Par ailleurs, mécaniquement, un modèle dans lequel on ajoute des covariables sera toujours plus fortement prédictif. Même si on ajoute des variables de corrélation nulle avec l’outcome, sur un échantillon fini, l’effet de cette variable sera légèrement positif ou négatif par hasard et cela améliorera la prédictivité (réduira la déviance) du modèle sur l’échantillon, avec pour effet de gonfler tous les odds ratio conditionnels.
Ainsi, deux odds ratio conditionnels à des choses différentes ne sont pas comparables. C’est pourquoi, au sens strict, on ne peut pas comparer un odds ratio dans un modèle avec une covariable à l’odds ratio de la même covariable dans un modèle avec une dizaine d’ajustements supplémentaires. De manière mécanique, l’odds ratio conditionnel à la dizaine de variables d’ajustement sera gonflé. En pratique, cela pose rarement problème en médecine, car les « facteurs pronostiques » sont généralement faiblement corrélés à l’outcome, conduisant à une inflation modeste des odds ratio conditionnels. Néanmoins, j’ai un beau cas d’école, pour lequel il existe une solution élégante. Le problème était d’évaluer les causes de la variance inter-établissement en terme de proportion de césariennes chez les femmes parturientes. La variance inter-établissement était exprimée comme l’odds ratio médian, interprétable comme la médiane des odds ratio entre deux établissements pris au hasard (le détail du calcul est un peu plus complexe et se basait sur la variance inter-établissement dans un modèle linéaire généralisé à effets mixtes, mais c’est approximativement ça). Or, les établissements n’ont pas la même population. Certains suivent des femmes enceintes à très bas risque alors que d’autres suivent des femmes à haut risque. La classification de Robson (doi:10.1016/j.jgyn.2015.02.001) définit 12 groupes de femmes, avec des risques extrêmement différents, allant de 2.1% pour les femmes multipares portant un singleton céphalique > 37 SA avec travail spontané, jusqu’à 82.4% pour les primipares avec présentation siège. Il était attendu que l’analyse brute montre un fort odds ratio inter-établissement médian, et qu’il s’atténue lors de l’ajustement sur le Robson. Au contraire, l’odds ratio inter-établissement médian a augmenté. Cela est explicable par le fait que des différences minimes dans les sous-groupes à très bas ou très haut risque (p.e. 2.1% -> 1% pour le sous-groupe à bas risque) s’expriment par des odds ratio conditionnels importants alors que l’effet marginal (20% -> 19%) s’exprime par un odds ratio bien plus modeste. Outre l’usage d’un modèle linéaire qui résout complètement le problème, on peut calculer dans chaque établissement un taux standardisé de césarienne en faisant la moyenne des taux des sous-groupes, pondérée par une fréquence de référence de ce sous-groupe de Robson. La fréquence de référence des sous-groupes peut se baser sur la littérature ou simplement être obtenu en poolant tous les établissements afin d’obtenir une population de référence représentative de l’ensemble des centres. Les odds ratio obtenus entre ces taux standardisés sont marginaux et sont alors comparables aux odds ratio d’un modèle non ajusté.
Enfin, il existe un problème à ne pas négliger : les modèles de régression logistiques dits « conditionnels » que l’on utilise en cas d’appariement, comme, par exemple dans les études en cross-over fournissent un odds ratio ininterprétable. Pour faire bref, ils fournissent une probabilité conditionnelle à une condition inobservable. Cela s’illustre bien en considérant le cas d’une maladie chronique stable pour laquelle on essaye un traitement symptomatique (p.e. traitement de la douleur dans la polyarthrite rhumatoïde) et pour laquelle un essai en cross-over est pertinent. Considérons pour critère de jugement, la réponse binaire au traitement, défini par une amélioration de la douleur. Considérons l’usage de deux traitements de nature très proche et tous deux assez inefficaces. On peut imaginer que 90% des sujets ne répondront à aucun des deux traitements, 8% des sujets répondront aux deux traitements, 1.5% des sujets répondront au traitement innovant mais pas au traitement de référence et 0.5% des sujets répondront au traitement de référence mais pas au traitement innovant. Dans ce contexte, les taux de réponses avec le traitement de référence et le traitement innovant sont respectivement de 8.5% et 9.5%, soit un différence absolue de pourcentage de 1% (nombre de sujets à traiter pour obtenir une réponse supplémentaire = 100) ou un odds ratio marginal à 1.13. L’odds ratio conditionnel au patient est égal à 3.0. En effet, cet odds ratio conditionnel au patient est calculé seulement sur les 2% de paires de mesures discordantes. Les 80% de paires concordantes négatives ne participent pas à l’estimation de l’odds ratio car les chances de réponses sont de 0% dans cette paire et tous les odds ratio ont la même vraisemblance . De même pour 8% de patients répondant aux deux traitements. La statistique est seulement calculée comme le rapport entre les discordances favorables au traitement innovant (1.5%) et les discordances favorables au traitement de référence (0.5%). Le rapport 1.5%/0.5% est égal à 3.0, l’odds ratio conditionnel au patient, ou encore, pour être plus clair, conditionnel au fait que le patient ait une réponse différente aux deux traitements. Cet odds ratio à 3.0 est extrêmement élevé par rapport à l’odds ratio marginal car la corrélation intra-patient est forte, ou, en d’autres termes, le facteur patient est fortement pronostique ! Le problème, c’est que ce facteur patient est inobservable tant qu’on n’a pas essayé les deux traitements. Si on considère que les patients se divisent en trois catégories : les rouges (80%), qui ne répondent à aucun des deux traitements, les verts (8%) qui répondent aux deux traitements et les oranges (2%) qui répondent à l’un mais pas à l’autre, alors l’odds ratio fourni s’applique au sous-groupe des patients oranges. Il est conditionnel à la couleur orange. Si la couleur était une variable clinique identifiable, alors il serait aisé d’orienter la prescription. Les patients rouges n’auraient aucun de ces deux traitements, les patients verts auraient l’un ou l’autre, à la préférence du médecin ou du patient et les patients oranges auraient en priorité le traitement innovant avec trois fois plus de chances de réponse que si on leur donnait le traitement de référence. Problème majeur : la couleur est inobservable, sauf à réaliser une période d’essai cross-over pour le patient qui conduirait à identifier d’emblée le sens de la différence et donc, à l’inutilité de la connaissance de cet odds ratio. En bref, l’odds ratio est conditionnel à une donnée inconnue.
Le problème décrit avec la régression logistique conditionnelle s’applique aussi aux régressions logistiques à effets mixtes dont les effets sont conditionnels aux effets aléatoires qui reposent sur des variables inobservables. Ces considérations n’existent pas dans les modèles linéaires à effets fixes ou à effets mixtes gaussiens parce que les effets conditionnels dans ces modèles sont égaux aux effets marginaux la fonction de lien étant l’identité. C’est pourquoi l’essai en cross-over imaginaire cité ci-dessus pourrait être analysé avec une estimation de la différence absolue de chances de réponse par la méthode de Student sur séries appariées. Cette méthode est asymptotiquement correcte et fournit un résultat pertinent et indépendant du degré de corrélation intra-paire.
Au final, rien n’est plus simple et interprétable que les résultats de modèles linéaires gaussiens. Les odds ratio conditionnels risquent d’être incorrectement interprétés comme des odds ratio marginaux alors qu’ils sont toujours gonflés de manière plus ou moins extrême selon que la condition explique plus ou moins fortement la variance de l’outcome. Les modèles logistiques dits ‘conditionnels’ sont à éviter car fournissent des résultats théoriquement ininterprétables, en pratique, fournissent des odds ratio interprétés à tort comme marginaux, et donc plus ou moins fortement biaisés.
Ce billet discute de l’ajustement dans les essais cliniques en randomisation individuelle. Il montre que l’ajustement statistique fait peu gagner en puissance, permet parfois de rectifier un défaut de randomisation, mais pose aussi des soucis non négligeables :
P-hacking plus ou moins important
Erreurs de manipulation du logiciel
Résultats théoriquement non biaisés mais ininterprétables (odds ratio conditionnel à une variable inobservée), en pratique interprétables mais biaisés (car interprétés comme odds ratio marginal ou conditionnel aux variables observées)
Résultats mal présentés (p.e. odds ratio conditionnel plutôt que différence absolue de risques)
Les hypothèses faites par les modèles (absences d’interactions, linéarité) sont fausses, et cela peut conduire à un biais dans les estimations
Une partie de ces problèmes disparaît ou s’atténue lorsqu’on utilise exclusivement le modèle linéaire gaussien, qu’on ajuste seulement sur la covariable pronostique majeure, qu’on s’assure que le protocole soit publié avec un paragraphe statistique très exhaustif.
Je conseille d’avoir conscience de ces problèmes afin d’utiliser avec parcimonie, et en toute connaissance de cause, les ajustements dans les essais cliniques en randomisation individuelle.
Introduction
Dans un essai clinique randomisé bien mené, il n’y a pas de facteur de confusion car l’allocation des traitements est aléatoire et donc indépendante de tout facteur pronostique ou pas. Un ajustement statistique sur des facteurs pronostiques est possible, avec ou sans stratification de la randomisation. Cet ajustement réduit les fluctuations d’échantillonnage explicables par les déséquilibres aléatoires entre les groupes sur ces facteurs pronostiques, ou, dans le cadre de la randomisation stratifiée, évite une surestimation des fluctuations d’échantillonnages, en excluant du calcul de la variance résiduelle, la variance ou plutôt la non-variance des facteurs pronostiques. Au final, cela augmente la puissance sans modifier le risque alpha. Comme dans l’évaluation de toute méthode statistique, deux questions doivent être posées : que gagne-t-on ? que perd-on ?
Que gagne-t-on ?
Le gain de puissance est maximal lorsque le facteur pronostique expliquant une grande partie de la variance de l’outcome. En pratique, cela veut dire que le facteur pronostic est fortement lié à l’outcome et a une grande variance. Pour un facteur pronostique binaire, l’idéal est une prévalence de 50% (forte variance) et que selon sa présence ou non, l’outcome soit très favorable ou défavorable.
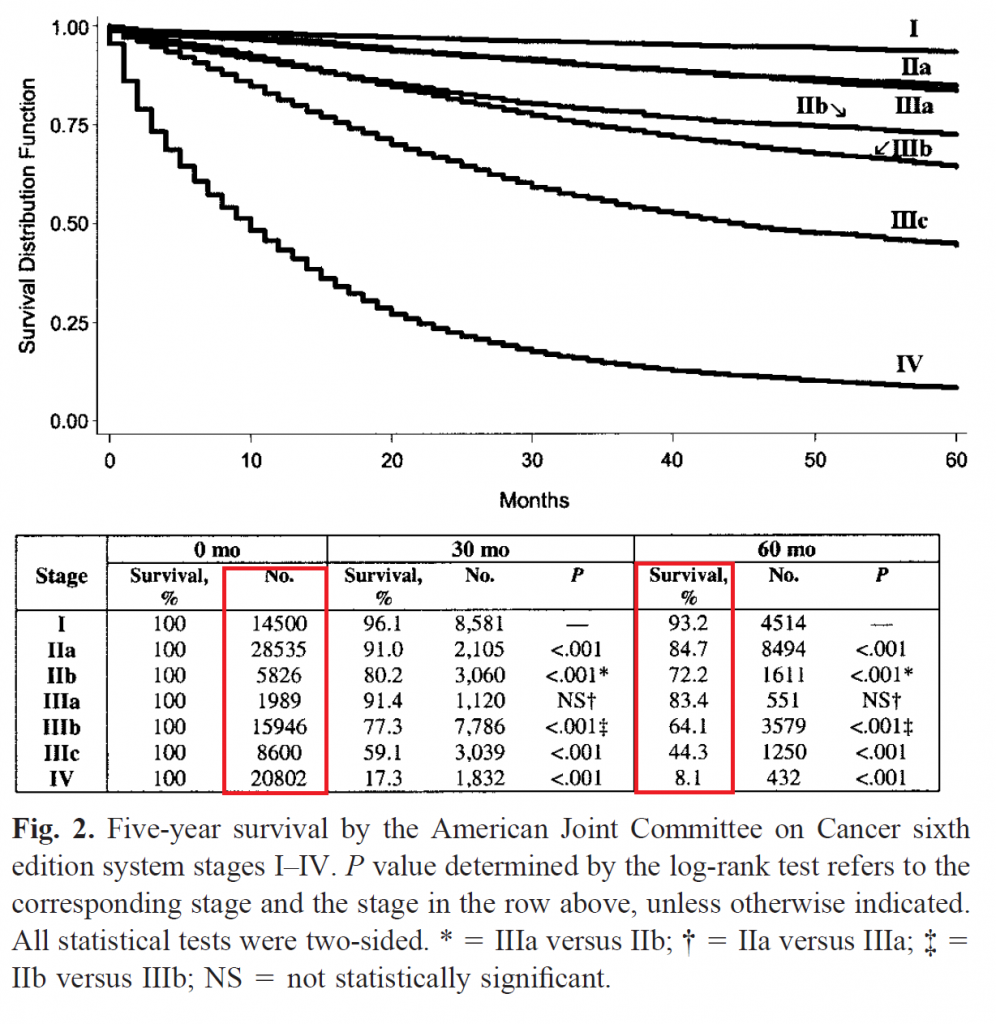
Nous prendrons comme référence, un cas d’école dans lequel le facteur pronostique est majeur et a une forte variance : le stade selon l’American Joint Committee on Cancer (AJCC) du cancer du colon, communément appelé stade TNM. Nous prenons comme référence la version 6 (O’Connell J, Maggard M, Ko C. Colon Cancer Survival Rates With the New American Joint Committee on Cancer Sixth Edition Staging. Journal of National Cancer Institute, Vol. 96, No. 19, October 6, 2004) car elle est en accès libre et dispose de tables de mortalité et de prévalences précises contrairement aux versions 7 et 8. La figure ci-dessous montre la distribution jointe du stade et de la mortalité à 5 ans.
Un facteur pronostique capable de séparer 8.1% de mortalité de 93.2%, ainsi que de nombreux risques intermédiaires, avec une variance aussi grande n’est pas commun dans les essais cliniques. Il faut noter qu’un essai clinique n’inclura pas des stades I et des stades IV. Le pronostic et le traitement de référence dépendant beaucoup du stade, un essai clinique inclura un seul stade (exemples : https://dx.doi.org/10.1056/NEJMoa1713709, https://dx.doi.org/10.1001/jamaoncol.2019.6486). Autrement, mélanger des patients ayant des stades très différents ouvre la porte à des interactions et la sélection d’un traitement bénéfique à un sous-groupe (p.e. stade III) mais nocif à un autre (p.e. stade II), sans que la distinction ne soit faite entre les deux stades. En bref, un tel facteur pronostic majeur ne devrait pas exister à l’intérieur d’un essai clinique. Considérons dans un premier scenario qu’il soit possible d’inclure des stades I (tumeur localisée ne dépassant pas la muqueuse) et des stades IV (tumeur avec métastases à distance) dans un même essai clinique.
Nous ne nous intéresserons pas aux modèles de survie basés sur les risques proportionnels, qui ont leur propres problèmes, mais plutôt considèrerons la mortalité binaire à 5 ans.
Le 1er scenario est basé sur une puissance à 80%, un risque alpha unilatéral à 2.5% (ou bilatéral à 5%) dans un modèle ajusté sur le stade tumoral (7 modalités -> 6 variables binaires recodées) en randomisation simple 1:1 dans un modèle linéaire gaussien pour une intervention correspondant à un odds ratio conditionnel à 0.50 (facteur protecteur) sur la mortalité à 5 ans. L’omission d’un ajustement est à l’origine d’une inflation de l’erreur type (standard error) par un facteur 1.32, correspondant à une réduction de la puissance de 80% à 56.4%. Pour atteindre la même puissance à 80%, il faut multiplier par 1.75 le nombre de sujets, passant de 519 sujets à 907 sujets. Le gain en terme de puissance est peu dépendant de l’effet du traitement. Un odds ratio à 0.25 conduit à une perte de puissance de 80% à 57.1% lors du non-ajustement alors qu’un odds ratio à 0.95 conduit à une perte de puissance de 80% à 57.4% lors du non-ajustement. Cela est dû au fait que la perte de puissance dépend seulement de la variance expliquée par le facteur pronostique, qui dépend de la prévalence et de la moyenne des outcomes les deux groupes étant poolés. Cette moyenne des outcomes dépend un peu de l’effet du traitement.
Le 1er scenario montre un intérêt important de l’ajustement mais n’est pas réaliste du tout. Aucun essai clinique n’inclurait des stades I à IV. Le 2ème scenario, encore loin de la réalité, est basé sur l’inclusion des stades I à III (tumeur très localisée, jusqu’à tumeur avec envahissement local important touchant les organes voisins et métastases ganglionnaires régionales nombreuses). Dans ce cas là, le non-ajustement (avec odds ratio à 0.50) conduit à une inflation de l’erreur type par 1.061, une réduction de la puissance de 80% à 75.2% ou une augmentation du nombre de sujets nécessaires de 12.5%. Comme dans le 1er scenario, l’effet du traitement n’a pas vraiment d’importance.
Le 3ème scenario est identique au second même une régression logistique est employée plutôt qu’une régression linéaire. La perte de puissance est presque la même que dans le modèle linéaire, à une différence absolue de puissance de 0.98% près. Par contre, le modèle logistique risque d’être mal interprété. En effet, le bénéfice de traiter tous les sujets avec le traitement innovant correspond à une réduction de la mortalité à 5 ans de 38.4% à 29.6% correspondant à une différence absolue de risque de 8.8% (meilleur manière d’en quantifier le bénéfice) ou à un odds ratio marginal de 0.675. L’odds ratio conditionnel au stade, par contre, est égal à 0.50 (soit 0.74 fois l’odds ratio marginal). C’est cet odds ratio conditionnel que les logiciels statistiques fournissent en sortie. Plus l’ajustement porte sur un facteur pronostique pertinent, plus l’odds ratio conditionnel s’écarte de l’odds ratio marginal. En l’absence d’ajustement, la régression logistique fournit l’odds ratio marginal, plus simple à interpréter car indépendant du nombre et de la nature des variables d’ajustement.
Le 4ème scenario, plus réaliste, consiste en l’ajustement sur le stade a, b ou c chez des patients tous en stade III, en conservant les autres paramètres (odds ratio conditionnel à 0.50, puissance à 80%, risque alpha à 2.5% unilatéral), dans un modèle linéaire gaussien. L’inflation de la variance due au non-ajustement est de 1.025 avec une perte de puissance de 1.97% (80% -> 78.03) ou un nombre de sujets nécessaires augmenté de 5.1%.
En bref, l’ajustement sur un facteur pronostique important et de forte variance, dans des conditions réalistes, peut faire gagner 2% de puissance soit 5% de sujets à recruter en moins. Il paraît déjà impossible de gagner 5% de puissance car il faudrait une hétérogénéité majeure de la population (stades I à III mélangés), ce qui conduirait à une réduction du nombre de sujets nécessaires d’environ 11%. On peut donc conclure que le gain, en nombre de sujets nécessaires sera toujours inférieur à 10%, et sera souvent situé en dessous de 5%. La présence de multiples facteurs pronostiques fournit rarement une capacité pronostique supérieure à celle du stade dans les cancers du colon. Par exemple, le Karnofsky n’est pas pris en compte dans le stade AJCC, probablement parce qu’il n’influence pas de manière majeure le pronostic.
Un second bénéfice, non négligeable de l’ajustement, c’est l’adaptation aux situations où l’essai clinique randomisé n’a pas été bien mené telle qu’une mauvaise randomisation ce qui peut arriver, par exemple, dans un essai en ouvert avec une procédure de randomisation par blocs de permutation de taille 4 stratifiée sur le centre, permettant rapidement au clinicien de deviner dans quel groupe un patient sera randomisé. Cela peut aussi parfois servir s’il y a eu un biais d’attrition différentiel. L’ajustement peut, dans une certaine mesure, rectifier le biais.
Que perd-on ?
L’ajustement dans les essais cliniques randomisés n’est pas dénué de risques. Même si les essais cliniques sont très souvent enregistrés dans des bases de données comme ClinicalTrials ou EudraCT, le résumé de protocole disponible précise le critère de jugement mais rarement l’analyse statistique. Les protocoles complets détaillant l’analyse statistique ne sont pas toujours disponibles. Si le protocole complet n’est pas disponible les ajustements peuvent être un facteur conscient ou inconscient de P-hacking. Il est fréquent de lancer un très grand nombre d’analyses de sensibilité dans les rapports d’analyse statistique et l’analyse qui permettra le passage du petit p en dessous de 0.05 risque d’être choisie comme analyse principale.
Même un protocole détaillant la méthode, laisse généralement des degrés de liberté.
Exemple : l’analyse principale sera faite en intention de traiter modifiée (exclusion des sujets randomisés sortis d’étude ou décédés avant d’avoir reçu la 1ère cure de traitement) et sera réalisée dans un modèle de Cox expliquant la survie sans progression ajustée sur le centre, l’âge, le Karnofsky, le stade tumoral et le type histologique. Les sujets perdus de vue seront censurés à la date des dernières nouvelles. Le hazard ratio de l’effet traitement sera comparé à la valeur 1 par un test bilatéral au seuil de significativité 5%.
Cela semble clair et précis, mais il manque un grand nombre de détails :
Par quelle méthode les ex-aequo seront-ils gérés : Efron, Breslow ou méthode exacte ?
Certains ajustements sont-ils réalisés par stratification (modèle de Cox conditionnel) ? Cela est parfois fait pour l’effet centre.
Le petit p est-il calculé par le test du rapport de vraisemblance, celui du score ou celui de Wald, ou encore un autre estimateur ?
Comment seront gérées les données manquantes sur les covariables, notamment le Karnofsky ?
Comment seront recodées les variables. L’âge, par exemple, peut être introduit sous forme d’une unique variable quantitative (effet log-linéaire), sous forme de polynôme, ou découpé en catégories à seuils prédéfinis (p.e. 18-60, 60-75, >75 ans) ou découpé en quantiles (quintiles, quartiles, tertiles).
Quel est le niveau de détail du stade ? Distingue-t-on le stade IIa du stade IIb ou regroupe-t-on ces deux modalités en stade II ?
Quel est le niveau de détail du type histologique ? En effet, on peut toujours sous-typer très précisément avec la biologie moléculaire, ou pas…
La variable de stade fait-elle référence au cTNM, au pTNM ? Est-ce le stade pré-traitement ou post-traitement néo-adjuvant ?
Comment sont gérées les valeurs aberrantes dans les covariables découvertes après le lever d’aveugle ?
Quelles conditions de validité seront testées ? Y aura-t-il des tests d’interaction ? Si oui, que fera-t-on si certains sont significatifs ?
Pour l’item 10, la réponse évidente (de mon point de vue), est la non prise en compte des interactions, mais il n’est pas sûr que ce soit le comportement de tous les statisticiens. De même, pour l’item 9, la réponse évidente est l’imputation de la valeur aberrante par la même méthode de gestion des données manquantes que les autres, évitant ainsi un biais de classement différentiel, mais j’ai souvent observé dans ma pratique de biostatisticien, le comportement correspondant à « corriger » la donnée à partir du dossier médical, en gardant la trace de la modification dans le script d’analyse.
En analyse non ajustée, il existe déjà un espace de liberté permettant du P-hacking (p.e. plusieurs estimateurs existent, la gestion des ex-aequo se pose toujours), mais ces possibilités explosent complètement en cas d’analyse ajustée. Enfin, il est toujours possible de ne pas respecter du tout le protocole, sans que ce soit pour autant visible dans l’article. J’ai déjà observé un essai clinique randomisé dont les résultats sont publiés dans le Lancet pour lequel le protocole spécifiait que l’analyse principale serait faite dans une régression logistique ajustée sur la sévérité de la maladie à baseline et d’autres variables qui ressortiraient dans l’analyse univariée. Au final, plusieurs techniques de pas-à-pas ont été utilisées et l’article a été publié avec un modèle log-binomial non ajusté sur la sévérité de la maladie à baseline mais ajusté sur un score clinique à baseline qui était ressorti dans les analyses pas-à-pas (basées sur la P-valeur puis sur l’AIC). Cet exemple ne prouve pas que ces écarts au protocole sont fréquents, mais qu’ils sont possibles. Comme le protocole n’a pas été publié, cela est invisible.
Au delà du P-hacking conscient ou inconscient, il existe le risque d’erreur d’analyse. La manipulation des outils statistiques n’est pas évidente et il est toujours possible de se tromper dans la programmation d’une imputation, d’un recodage de variable ou de l’estimation d’un modèle. Plus le nombre de variables impliquées est grand et plus le risque d’erreur est, a priori, élevé. Je ne dispose pas de données à ce jour pour évaluer la fréquence ou l’ampleur de ce problème.
Il apparaît aussi le problème du choix du modèle, pour lequel il est aisé de faire le mauvais choix. Sur un critère de jugement binaire, appliquera-t-on un modèle de régression logistique (logit-binomial), un modèle log-binomial, un modèle identité-binomial ou un modèle linéaire gaussien ? Si on ajuste sur l’effet centre, va-t-on utiliser les équations d’estimation généralisées (GEE), un modèle linéaire généralisé à effets mixtes (GLMM) ou un modèle linéaire généralisé à effet fixe ? Le problème, c’est que l’analyse univariée fournira aisément un résultat pertinent, alors que peu des modèles sus-cités fournissent des résultats aussi robustes. Une régression logistique fournit un odds ratio conditionnel ininterprétable dans un GLMM car conditionnel à des variables inobservables. Une régression logistique ou un modèle log-binomial fournit un odds ratio conditionnel rarement converti en effet marginal. Comme vu au-dessus, il est « gonflé » par rapport à l’effet marginal. Ensuite, la régression logistique comme la log-binomiale fournissent des statistiques relatives (odds ratio ou rapports de proportions) qui reflètent bien mal le bénéfice réel du traitement dans un essai clinique randomisé. En effet, un risque relatif à 0.80 sur un risque touchant la moitié des sujets correspond à une réduction de 10% du risque absolu, soit seulement 10 sujets à traiter pour éviter un événement. Par contre, le même risque relatif appliqué à un risque extrêmement rare tel que 0.1%, obligera à traiter 5000 personnes pour éviter un événement. C’est pour cela, par exemple, qu’une antibioproxphylaxie dans des chirurgies à très haut risque infectieux se justifie alors qu’elle serait futile dans des opérations à très bas risque.
Le modèle identité-binomial semble pertinent, évaluant directement une différence absolue de risque, mais celui-ci montre ses limites lorsque des interactions apparaissent dans le modèle. Comment estimer le bénéfice net d’un traitement lorsque la différence absolue de risque diffère selon le sous-groupe ? En présence d’une interaction quantitative, c’est l’effet marginal qui nous permet d’évaluer le bénéfice net. Si 10% des sujets (mauvais pronostic) voient leur risque d’évolution défavorable baisser de 30% alors que 90% des sujets (bon pronostic) voient leur risque d’évolution défavorable baisser de 20% alors en donnant le traitement innovant à l’ensemble des sujets ont réduit de 0.10×0.30 + 0.90×0.20 = 21%. Un modèle linéaire non ajusté estimera, sans biais, les 21% de bénéfice. Un modèle linéaire gaussien ajusté estimera aussi, sans biais, les 21% de bénéfice car l’homoscédasticité supposée par le modèle conduira à une statistique ajustée égale à la des sous-groupes pondérée par l’effectif du sous-groupe. Cela est une propriété de la méthode d’estimation des moindres carrés. Le modèle identité-binomial, par contre, supposera que la variance est plus faible pour les pourcentages proches de 0% ou de 100% et donnera donc un poids plus fort au sous-groupe correspondant. En caricaturant volontairement, si la population est composée d’un groupe de 80% de patients ayant un risque à 99% (réduit de à 89% par le traitement, soit -10%) et 20% de patients ayant un risque à 50% (réduit à 49% par le traitement, soit -1%), alors le modèle identité-binomial ajusté sur le facteur pronostique estimera le bénéfice du traitement à -6.2% (espérance de l’estimateur) contre -2.8% pour un modèle identité-binomial non ajusté ou un modèle linéaire gaussien ajusté ou non ajusté. Le vrai bénéfice, si on donne le traitement à tout le monde, correspond à cette seconde estimation.
Il est illusoire de penser que les interactions quantitatives n’existent pas. Déjà, par principe, s’il n’existe pas d’interaction quantitative dans un modèle logistique, alors il en existera dans le modèle linéaire et vice versa. Les deux modèles sont fondamentalement contradictoires (sauf s’ils contiennent tous deux tous les termes d’interactions possibles). Les autres modèles sont aussi contradictoires : log-binomial, probit, etc. La stratégie consistant à tester les interactions et à accepter l’hypothèse nulle d’absence d’interaction par défaut de preuve du contraire, serait inepte à un bayésien qui sait que la probabilité a priori d’interaction est à peu près égale à 100%. C’est pourquoi, il est préférable de construire des statistiques « robustes » aux interactions quantitatives. Pour cela, il est possible d’utiliser un modèle linéaire gaussien ou identité-binomial non ajusté ou un modèle linéaire gaussien ajusté. Il est aussi possible de se baser sur un modèle AVEC interactions dès le départ, puis de reconstruire la statistique marginale en calculant la moyenne des effets dans chacun des sous-groupe pondérée par la taille du sous-groupe. Comme la covariance entre les sous-groupes devient nulle lorsqu’on utilise un modèle avec des interactions complètes, le calcul n’est pas super compliqué. Le choix du modèle n’a alors plus d’importance puisqu’on estime directement les effets moyens en sous-groupes. En présence de variable pronostique quantitative, il n’y a pas de solution de codage parfaite parce qu’on ne peut pas modéliser la nature de la relation de manière exacte. Le problème disparaît si on n’ajuste pas du tout ou si on découpe la variable quantitative en tranches (ajustement partiel).
En cas d’interaction qualitative, les problèmes décrits ci-dessus sont exacerbés. C’est probablement un phénomène rare, heureusement, mais il est préférable de se comporter aussi bien que possible s’il advenait. Dans le cas d’une interaction qualitative, le modèle identité-binomial est susceptible de montrer un effet opposé à l’effet marginal réel ! Les interactions qualitatives sont problématiques dans tous les cas parce que le traitement est bénéfique à un sous-groupe identifiable de patients alors qu’il est nocif à un autre sous-groupe identifiable de patients. Malheureusement, la puissance statistique nécessaire à la détection de ce phénomène est loin d’être satisfaisante dans la plupart des études. Cela est d’autant plus vrai que les sous-groupes sont déséquilibrés. C’est pourquoi on évitera, dès le départ, par des critères d’inclusion judicieux, d’avoir une population trop hétérogène avec une probabilité d’interaction forte. Cela justifie, par exemple, qu’on évite de mélanger des enfants et des adultes dans les évaluations de traitements psychiatriques. Néanmoins, dans le cas où on se fait avoir, c’est-à-dire, où il existe une interaction qualitative sur une variable qu’on n’avait pas anticipé et pour laquelle la puissance est totalement insuffisante pour retrouver une tendance dans une analyse en sous-groupes, alors on devra « vivre » avec et donner le traitement innovant à tout le monde. C’est la même chose avec les variables d’interaction inconnues (p.e. dosage biologique encore inconnu) : en l’absence de connaissance, on évalue le bénéfice global. C’est là que l’effet marginal directement estimé par le modèle linéaire gaussien reflètera le bénéfice pragmatique. Empiriquement, il semblerait que le modèle de régression logistique ne puisse pas conduire à l’erreur de 3ème espèce (effet significatif dans le sens opposé à l’effet réel) là où le probit-binomial, l’identité-binomial et le log-binomial peuvent. Il semblerait qu’en présence d’interaction, même si elle n’est pas prise en compte dans le modèle logistique, l’effet marginal prédit par le modèle logistique est correct (identique au modèle linéaire ou au modèle non ajusté) dans le cas d’un ajustement sur une covariable catégorielle.
Il existe aussi le problème de non convergence très fréquent dans le modèle log-binomial lorsqu’on ajuste sur une variable quantitative ou sur de nombreuses variables binaires. Cela est dû au fait que le modèle est grossièrement faux, conduisant à des prédictions dépassant le risque de 1. Il y a de nombreuses interactions quantitatives ou défauts de log-linéarité dans ces modèles et comme vu au-dessus, il n’est plus possible d’estimer un effet conditionnel ou marginal de manière fiable. Par ailleurs, que faire en cas de non convergence ? Fournir un effet non ajusté ? Cela paraît raisonnable, mais la procédure en deux étapes entraîne une erreur statistique mal caractérisée. C’est par exemple, lorsque l’effet d’une covariable est surestimé, que le défaut de convergence apparaît, et dans ce cas précis, on supprime l’ajustement, supposant forçant son effet est à zéro et créant une fluctuation d’échantillonnage discrète sur la statistique principale. En bref, en ajoutant un seul sujet, on peut changer l’estimation principale de manière principale. Sous l’hypothèse d’absence d’effet de la variable principale, cela entraîne une erreur mais pas un biais car ça n’a aucune raison d’avantager un groupe que l’autre. Sous l’hypothèse d’existence d’un effet, cela pourrait peut-être engendrer un biais minime, car il y a à peu près une chance sur deux que cela fasse fluctuer la statistique principale dans un sens et une chance sur deux que ça la fasse fluctuer dans l’autre étant donné que la corrélation entre la covariable et la variable principale est totalement aléatoire de moyenne que la variable
Il existe aussi le problème de non convergence très fréquent dans le modèle log-binomial lorsqu’on ajuste sur une variable quantitative ou sur de nombreuses variables binaires. Cela est dû au fait que le modèle est grossièrement faux, conduisant à des prédictions dépassant le risque de 1. Il y a de nombreuses interactions quantitatives ou défauts de log-linéarité dans ces modèles et comme vu au-dessus, il n’est plus possible d’estimer un effet conditionnel ou marginal de manière fiable. Par ailleurs, que faire en cas de non convergence ? Fournir un effet non ajusté ? Cela paraît raisonnable, mais la procédure en deux étapes entraîne une erreur statistique mal caractérisée. C’est par exemple, lorsque l’effet d’une covariable est surestimé, que le défaut de convergence apparaît, et dans ce cas précis, on supprime l’ajustement, supposant forçant son effet est à zéro et créant une fluctuation d’échantillonnage discrète sur la statistique principale. En bref, en ajoutant un seul sujet, on peut changer l’estimation principale de manière principale. Sous l’hypothèse d’absence d’effet de la variable principale, cela entraîne une erreur mais pas un biais car ça n’a aucune raison d’avantager un groupe que l’autre. Sous l’hypothèse d’existence d’un effet, cela pourrait peut-être engendrer un biais minime, car il y a à peu près une chance sur deux que cela fasse fluctuer la statistique principale dans un sens et une chance sur deux que ça la fasse fluctuer dans l’autre étant donné que la corrélation entre la covariable et la variable principale est totalement aléatoire et d’espérance nulle. Difficile de dire si l’erreur aléatoire augmente un peu le risque alpha ou le diminue un peu. En tout cas, l’erreur aléatoire ne suit pas une loi normale.
Une autre option, en cas de non-convergence d’un modèle log-binomial, c’est d’utiliser un modèle tout autre à ce moment là, tel qu’un modèle logistique. Cela est problématique parce que la statistique fournie n’est plus comparable. Les fluctuations d’échantillonnage fréquentistes deviennent presque ininterprétables puisque la base de la statistique fréquentiste, c’est qu’un même protocole conduit à une même statistique.
Le modèle identité-binomial a des défauts de convergence aussi, même s’ils sont beaucoup plus rares que dans le modèle log-binomial. L’usage d’un modèle linéaire gaussien avec des ajustements parcimonieux (une ou deux variables pronostiques majeures, les autres étant généralement futiles) évite le problème.
Le problème de non-convergence est aussi une problématique à prendre en compte dans le protocole, qui doit préciser l’usage de modèles de secours. Autrement, cela pourrait être un facteur de P-hacking ou de non reproductibilité des analyses.
Synthèse
Nous avons vu qu’un ajustement statistique dans un essai clinique randomisé permet de recruter 5% de patients en moins en cas de facteur pronostic important et que ce chiffre peut monter jusqu’à 10% dans les conditions ou un facteur pronostic absolument majeur existe (en mélangeant les stades I à III du cancer du colon dans un même essai clinique par exemple). Il existe aussi un bénéfice de rectification des biais dû à une étude mal conduite, telle qu’une triche avec la randomisation.
Par contre, l’ajustement pose des problèmes de P-hacking et a de nombreuses limites d’interprétation à moins qu’il ne soit réalisé de manière très rigoureuse:
Précision exacte de la méthode de traitement des données manquantes des covariables
Précision du paramétrage exact du modèle, tel que l’estimateur
Précision du codage exact des covariables rentrées dans le modèle
Usage d’un modèle linéaire gaussien pour une variable binaire plutôt que les régressions logistiques, log-binomiales ou identité-binomiales
Publication du protocole avec le plan d’analyse statistique le plus tôt possible !
Cela est plus facile à réaliser si vous ajustez sur peu de covariables et vous concentrez seulement sur une ou deux covariables pronostiques majeures, pour laquelle la qualité de la donnée est bonne (donnée de routine bien renseignée). S’il y a plusieurs covariables, ajoutez les termes d’interaction afin d’éviter les problèmes de fausses hypothèses.
Si vous êtes méthodologiste, je vous invite à être extrêmement explicite dans le protocole même pour un modèle non ajusté. Je vous conseille de ne faire un ajustement qu’à condition (i) de vous assurer que le protocole sera publié et (ii) qu’il existe un facteur pronostique majeur.
Si vous lisez un article, je vous conseille de toujours essayer d’obtenir le protocole, et de vérifier, s’il existe, qu’il a été respecté, notamment sur le choix du critère de jugement principal et le choix de l’analyse principale. Si le protocole n’est pas publié ou qu’il est incomplet, méfiez vous des analyses ajustées.
Les randomisations par blocs de permutation, de tailles fixe ou aléatoire, sont très utilisées dans les essais cliniques randomisés, qu’ils soient en aveugles ou en ouverts. Il arrive souvent que la randomisation soit stratifiée sur un ou plusieurs facteurs, tels que le centre ou des facteurs pronostiques, multipliant ainsi les listes de randomisation. D’un point de vue théorique, ces méthodes réduisent les déséquilibres en effectif entre les groupes (et sous-groupes) et améliorent la stabilité des estimateurs, conduisant à une meilleure puissance statistique. Malheureusement, il semblerait que le gain de puissance soit minime et qu’il y ait un risque de biais d’allocation, dans les études en ouvert.
Nous décrirons d’abord, un point de vue épistémologique, puis fournirons les détails statistiques permettant d’apprécier le gain de puissance et la perte de validité
Point de vue épistémologique
Avant d’utiliser la randomisation, d’autres moyens d’allocation pseudo-aléatoire de traitements avaient été utilisés, notamment l’inclusion dans le groupe A les jours pairs du calendrier et l’inclusion dans le groupe B les jours impairs. Cette méthode, semble, à première vue, fournir des groupes comparables, puisqu’il n’y a aucune raison que les caractéristiques du patient puissent être corrélées à la parité du jour. Néanmoins, cette méthode avait le défaut d’être prévisible. En situation où de grands espoirs sont portés sur le nouveau traitement, notamment lorsque le traitement de référence est peu efficace, la maladie sévère, et les bénéfices potentiellement perçus du traitement innovants sont importants, alors l’investigateur est susceptible d’utiliser cette information pour forcer le pseudo-hasard dans le sens qu’il souhaite. Souhaitant faire bénéficier du traitement innovant le patient dont le pronostic sous traitement de référence est péjoratif, il pourra décaler la consultation afin de s’assurer que celle-ci tombe un jour favorable. Dès lors qu’une information partielle ou totale sur le groupe dans lequel le patient se retrouverait existe avant l’inclusion, le destin peut être modifié par la volonté humaine. Dans le pire des cas, l’inclusion est différée et une nouvelle consultation est programmée. Si ce destin est modifié de manière répétée, un déséquilibre apparaît entre les deux groupes : il ne sont plus comparables. Le biais d’indication des études observationnelles ré-apparaît partiellement ou pleinement.
Le seul moyen d’éviter ce biais est la parfaite imprévisibilité de la séquence d’allocation. La randomisation, c’est-à-dire l’allocation au hasard, garantit cette imprévisibilité car le hasard n’est corrélé à aucune variable observée ou inobservée. La randomisation par blocs, ne répond pas à cette définition de hasard. Elle a une auto-corrélation négative et est donc prévisible dès lors que la randomisation est ouverte. Un investigateur incluant 4 patients d’affilée dans le même groupe peu parier que le prochain patient sera alloué dans l’autre groupe. L’algorithme est tellement simple, que son exploitation peut être inconsciente.
Gain de puissance
Le code R ci-dessous calcule la perte de puissance due à un déséquilibre des groupes en randomisation 1:1 avec une unique liste de randomisation:
power_for_n1=function(power_for_n, sig.level, n1, N) {
# On recrute N sujets en randomisation simple
# On observe n1 sujets dans le groupe A
# On veut connaître la puissance (conditionnelle à n1)
df = (N-2)
# espérance de la statistique T de Student pour n = N/2
# (randomisation par blocs -> équilibre parfait des groupes)
T_for_n = qt(power_for_n,df=df) + qt(1-sig.level/2,df=df)
# taille de n2 sachant que n1+n2 = N
n2 = N-n1
n = N/2
# espérance de la statistique T de Student
# pour la randomisation simple (conditionnelle à n1)
T_for_n1 = T_for_n * sqrt((1/n + 1/n)/(1/n1 + 1/n2))
# puissance pour la randomisation simple (conditionnelle à n1)
pt(T_for_n1 - qt(1-sig.level/2,df=df), df=df)
}
# n1 suit une loi binomiale
power_for_nrandom=function(power_for_n, sig.level=0.05, N=100) {
x = 1:(N-1) # valeurs possibles pour n1
d = dbinom(x, N, 0.50) # probabilités associées
p = power_for_n1(power_for_n, sig.level, x, N) # puissances conditionnelles
sum (d * p) # puissance inconditionnelle
}
On peut ainsi calculer qu’une randomisation simple 1:1 avec une seule liste, fait passer une puissance de 80% à 79.8% (réduction de 0.2%) pour une étude avec 200 sujets en tout (~100 par groupe).
Biais d’allocation
Un coût non négligeable existe dans les études en ouvert : le groupe auquel va appartenir le prochain patient potentiellement incluable est partiellement prévisible. Par exemple, dans une randomisation avec blocs de permutation de taille 4, il ne peut jamais y avoir plus de 4 patients d’affilée dans le même groupe. Cela est possible qu’un bloc est ‘0011’ avec le bloc suivant à ‘1100’. Dans une étude monocentrique, un investigateur qui a inclus 4 fois d’affilée des patients dans le même groupe sait alors dans quel groupe le prochain patient sera affecté. Dans une moindre mesure, si trois patients sont affectés d’affilée dans le même groupe, il y a 70.8% de chances que le prochain patient soit affecté à l’autre groupe. Le code ci-dessous simule ce scenario:
set.seed(2020)
v=as.vector(replicate(1000000,sample(c(1,1,0,0),replace=F)))
count_succ=function(v) {
w = rep(0, length(v))
for(i in 2:length(v)) {
if (v[i] == 1) {
w[i] = w[i-1]+1
}
}
w
}
w=count_succ(v)
frst=(1:(length(v)-1))
nxt=(2:length(v))
mean((v[nxt])[w[frst]])
L’investigateur n’a même pas besoin de compter précisément les patients. Il lui suffit de se souvenir des derniers patients pour anticiper le prochain. Ce problème existe même en cas de bloc de taille aléatoire, même si le calcul de probabilité n’est plus exact. Dans les essais multicentriques, avec randomisation centralisée, le problème est réduit par le fait que plusieurs investigateurs peuvent inclure sans communiquer. Même si un investigateur peut avoir inclus 4 patients d’affilée dans le même bras, il est possible que d’autres investigateurs, entre temps, aient inclus des patients, de telle sorte que son information sur la séquence aléatoire est brouillée. Néanmoins, il est fréquent de stratifier la randomisation sur le centre, créant ainsi une liste différente pour chaque centre et laissant donc à l’investigateur autant d’information que dans un essai monocentrique. La seule randomisation qui garantisse des allocations indépendantes, c’est la randomisation simple ! Autrement, l’information disponible sur le groupe de randomisation dans lequel les premiers patients ont été alloués (accessibles dans un essai en ouvert) permet d’obtenir de l’information sur les patients suivants.
C’est pourquoi, au moins dans les essais ouverts, je conseille d’utiliser la randomisation simple. Le risque de biais est ainsi diminué alors que la puissance n’est presque pas abaissée.
Limites du raisonnement
De même que l’évaluation en aveugle, l’aveugle patient et l’aveugle investigateur, la randomisation n’est qu’un outil de rigueur méthodologique qui n’est pas toujours indispensable. L’article Impact of blinding on estimated treatment effects in randomised clinical trials: meta-epidemiological study ne montre pas une différence majeure (même s’il existe une incertitude non négligeable) d’effet selon la rigueur méthodologie des essais cliniques randomisés. Néanmoins, le cas moyen ne décrit pas le cas particulier. Il me paraît indispensable d’être rigoureux dans les contextes où les investigateurs perdent leur neutralité, comme par exemple, en ce moment (avril 2020) pour l’évaluation thérapeutique de l’hydroxychloroquine dans le traitement du COVID-19. Dans ce cas, la randomisation ouverte est à éviter. La randomisation en double aveugle préservant le secret de la séquence d’allocation, la randomisation par blocs n’est alors pas aberrante.